Samenvatting
In deze richtlijn wordt onderscheid gemaakt tussen de variantvorm Creutzfeldt-Jakob (vCJD) en de overige vormen van Creutzfeldt-Jakob (CJD): de sporadische, genetische en iatrogene vorm, deze worden in de richtlijn aangeduid als CJD.
Verwekker: Afwijkende prioneiwitten; lichaamseigen eiwitten met een afwijkende vorm
Incubatieperiode CJD: Variërend van 1 tot meer dan 40 jaar
Incubatieperiode vCJD: Waarschijnlijk gemiddeld 12 jaar (6 tot 18 jaar)
Besmettingsweg CJD: Via besmet weefsel of besmette hormoonpreparaten (zeldzaam)
Besmettingsweg vCJD: Oraal via voedingsmiddelen besmet met Boviene Spongiforme Encefalopathie (BSE), met name hersen- en zenuwweefsel van rund, zeldzaam is vCJD overgedragen via bloedtransfusie
Besmettelijke periode: Verschilt per soort weefsel
Maatregelen: Meldingsplicht groep C (let op: procedure verschillend van andere meldingsplichtige ziekten; brononderzoek vanuit ziekenhuis)
Symptomen CJD: Snel verlopende dementie, myoklonie, ataxie, piramidale - en extrapiramidale verschijnselen, corticale blindheid en (in eindstadium) akinetisch mutisme
Symptomen vCJD: Vooral psychiatrische stoornissen en pijnlijke gevoelsstoornissen en cerebellaire ataxie
blok
Deze richtlijn is ontwikkeld voor zorgprofessionals werkzaam binnen de infectieziektebestrijding. De primaire doelgroepen zijn GGD- en LCI-professionals. Deze richtlijn bevat adviezen, taken en verantwoordelijkheden en vormt een basis voor het nemen van geïnformeerde beslissingen en het maken van beleid in de praktijk. Voor meer informatie zie Ontwikkeling LCI-richtlijnen.
Vaststelling (Landelijk Overleg Infectieziektebestrijding): 23 januari 2024. Publicatie: 20 februari 2024.
- 26-01-2026: Per abuis ontbrekende paragraaf (Besmettingsweg (variant Creutzfeldt-Jakob Disease)) teruggeplaatst.
- 11-07-2025: Aanpassing bij Preventie vanwege het vervallen van de preventieve maatregel (per 7 april 2025) om bloeddonoren uit te sluiten die tussen 1 januari 1980 - 31 december 1996 zes maanden of langer in het Verenigd Koninkrijk verbleven. Sanquin heeft een risicobeoordeling gedaan en acht deze maatregel niet meer nodig.
- 18-03-2025: Nieuwe indeling in achtergrondinformatie en een richtlijndeel (met de hoofdstukken Diagnostiek, Preventie en Maatregelen en waar van toepassing arbeidsrelevante aanvullingen en veterinaire informatie). Historie is ondergebracht in andere hoofdstukken en ten dele vervallen.
- 03-10-2024: Het uitsluiten van bloeddonoren die vanaf 1980 zelf een bloedtransfusie hebben ontvangen, is komen te vervallen wegens een beleidswijziging in overeenstemming met Sanquin per 1 oktober 2024.
- 20-02-2024: De bijlage Creutzfeldt-Jakob - Extramurale procedures bij de melding vervangt het draaiboek extramurale procedures. De bijlage is in twee delen gesplitst: na de meldingsplicht wordt eerst ingegaan op de procedure van CJD, gevolgd door de procedure van vCJD. Stroomschema's verduidelijken de rollen en taken van de verschillende instanties in de routes van diagnostiek, melden en advies. Vaststelling LOI: 23 januari 2024 20-02-2024: Nieuwe versie van de richtlijn en draaiboek. Deze richtlijn is herzien door Ayla Hesp, (Landelijke Coördinatie Infectieziektebestrijding), (Rijksinstituut voor Volksgezondheid en Milieu) in afstemming met Erasmus (Medisch Centrum), Landelijke Registratie Prionziekten. Arts M&G Monica Wong heeft het hoofdstuk Maatregelen begeleid, evenals de Bijlage extramurale procedures bij melding van de ziekte van Creutzfeldt-Jakob. Het hoofdstuk Diagnostiek is geschreven door Charlotte Menart, Amber Yaqub en Arfan Ikram (Landelijke Registratie Prionziekten, Erasmus MC) en aangevuld door Marcel Verbeek (Radboud (Universitair Medisch Centrum Utrecht)) en Annemieke Rozemuller (van het voormalig prionlab UMCU). De volgende deskundigen hebben het concept van de richtlijn van commentaar voorzien: Charlotte Menart, Amber Yaqub en Arfan Ikram (Erasmus MC), Marcel Verbeek (Radboud UMC), Annemieke Rozemuller en Evelien Lemstra (Amsterdam UMC), Lucien van Keulen (Wageningen Bioveterinary Research), Roan Pijnacker (EPI, RIVM), Corien Swaan en Klaartje Weijdema (LCI, RIVM).
- De RT-QuIC test is toegevoegd aan de meldingscriteria, zie het hoofdstuk Maatregelen. In de praktijk worden obducties zelden meer gedaan, in plaats daarvan wordt nu vaak een RT-QuIC test gedaan om de diagnose te stellen.
- Verder is de richtlijntekst geüpdatet ten aanzien van de naamgeving: de term 'klassieke vorm' is vervangen door 'sporadische vorm' omdat de term 'klassieke vorm' tegenwoordig niet meer gebruikt. Nieuwe inzichten uit wetenschappelijke literatuur zijn toegevoegd en het hoofdstuk Epidemiologie is aangevuld met de meest recente gevallen in binnen- en buitenland.
Achtergronden
Verwekker
De ziekte van Creutzfeldt-Jakob (CJD) wordt veroorzaakt door prioneiwitten die anders gevouwen zijn. Deze verkeerd gevouwen prioneiwitten hebben een rol in de spongiforme encefalopathieën (sponsvormige hersenafwijkingen) (Prusiner, 1998). CJD hoort bij deze groep progressieve en ongeneeslijke neurodegeneratieve aandoeningen. De fysiologische functie van het gezonde prioneiwit in cellen is niet helemaal duidelijk. Prioneiwit kan zowel schadelijk zijn als een beschermende functie hebben (Carlson & Prusiner 2021, Corbett 2020, Kovač & Čurin Šerbec 2022, Laurén 2009, Matamoros-Angles 2023).
De ziekte van Creutzfeldt-Jakob (CJD) wordt onderverdeeld in vier verschillende vormen:
Sporadisch
Dit is de meest voorkomende vorm van CJD. Deze vorm wordt sporadisch genoemd, omdat de oorzaak tot nu toe onbekend is. Bij deze vorm zijn er geen familieleden met de ziekte en is er geen aanwijsbare overdraagbare oorzaak (Karamujić-Čomić 2022). Voorheen werd deze vorm vaak aangeduid als de 'klassieke vorm'.
Genetisch
Bij deze vorm is er een erfelijke oorzaak, zoals een mutatie in het priongen (PRNP), het gen dat codeert voor het prioneiwit. De PRNP-genmutaties leiden ook tot andere prionziekten. Binnen de genetische prionziekten worden naast CJD ook de ziekte van Gerstmann-Straussler-Scheinker (GSS) en de fatale familiaire insomnia (FFI) onderscheiden. Vanuit het oogpunt van de infectieziektebestrijding worden deze ziektebeelden hetzelfde behandeld als sporadische CJD.
Iatrogeen
Als CJD ontstaat door instrumentarium dat is besmet met prionen, spreken we van de iatrogene vorm. Dit is zeldzaam: in de periode 1993-2021 zijn slechts zes patiënten met deze vorm gezien in Nederland (Karamujić-Čomić 2022). Het gaat om personen die in de jaren zeventig en tachtig zijn behandeld met menselijk groeihormoon of een hersenvliestransplantatie hebben ondergaan.
Variant (vCJD)
vCJD is de aanduiding voor de vorm die van dier-op-mens wordt overgedragen. Het wordt veroorzaakt door consumptie van producten van dieren met boviene spongiforme encefalopathie (BSE, 'gekkekoeienziekte') (Brandel 2009, Karamujić-Čomić 2022). Tussen 2005 en 2008 is deze diagnose in Nederland drie keer gesteld. Ook is vCJD incidenteel mens-op-mens overgedragen via bloedtransfusie in het Verenigd Koninkrijk (Hewitt 2006).
CJD is een zeldzame aandoening (iets meer dan 1 op de miljoen per miljoen inwoners per jaar), waarbij sporadische CJD de meest voorkomende vorm is (85-95% van de gevallen is sporadisch). Van het totale aantal CJD-gevallen is 5-15% genetische CJD. Variantvorm CJD en iatrogene CJD betreffen samen slechts 1% van de gevallen. Beide laatste vormen van CJD zijn in Nederland al meer dan 10 jaar niet meer voorgekomen.
In deze richtlijn wordt de variantvorm CJD onderscheiden van de overige vormen vanwege de bijzondere besmettingsroute bij vCJD, daarom is er ander beleid voor vCJD. Voor de overige drie vormen is het beleid hetzelfde (namelijk als bij de sporadische vorm). Een belangrijke reden daarvoor is het zeldzame karakter van de ziekte van Creutzfeldt-Jakob. In deze richtlijn worden de sporadische, genetische en iatrogene vorm daarom gezamenlijk aangeduid als CJD, de variantvorm wordt aangeduid als vCJD.
Epidemiologie
Verspreiding in de wereld
CJD
De incidentie van sporadische CJD is in Nederland en wereldwijd de afgelopen decennia iets toegenomen. Deze toename lijkt onder andere veroorzaakt door toegenomen bekendheid met de ziekte, verbeterde diagnostiek en verbeterde surveillance (Rhoads 2020). Het voorkomen van genetische CJD is 30-100 keer hoger in bepaalde delen van de wereld: Noord-Afrika, Israël, Italië en Slowakije (Ladogana 2005).
vCJD
De vCJD-epidemie heeft zich met name afgespeeld in het Verenigd Koninkrijk in de jaren tachtig en negentig, gerelateerd aan de toen grootschalige epidemie van BSE. Tot op heden worden de meeste gevallen van vCJD dan ook gemeld vanuit het Verenigd Koninkrijk (in oktober 2021 waren er 232 gevallen wereldwijd, waarvan 178 uit het Verenigd Koninkrijk (Figuur 1) (McManus 2022). Na het Verenigd Koninkrijk heeft Frankrijk de meeste gevallen gehad (28). Daarnaast zijn er enkele gevallen geweest in onder meer Spanje (5), Ierland (4) en de Verenigde Staten (4), Italië (3) en Nederland (3) (zie Figuur 1: Gerapporteerde gevallen van vCJD wereldwijd). Individuele gevallen in Japan en Taiwan hebben zich voorgedaan bij personen die ten tijde van de BSE-crisis gedurende ten minste enkele weken in het Verenigd Koninkrijk verbleven.
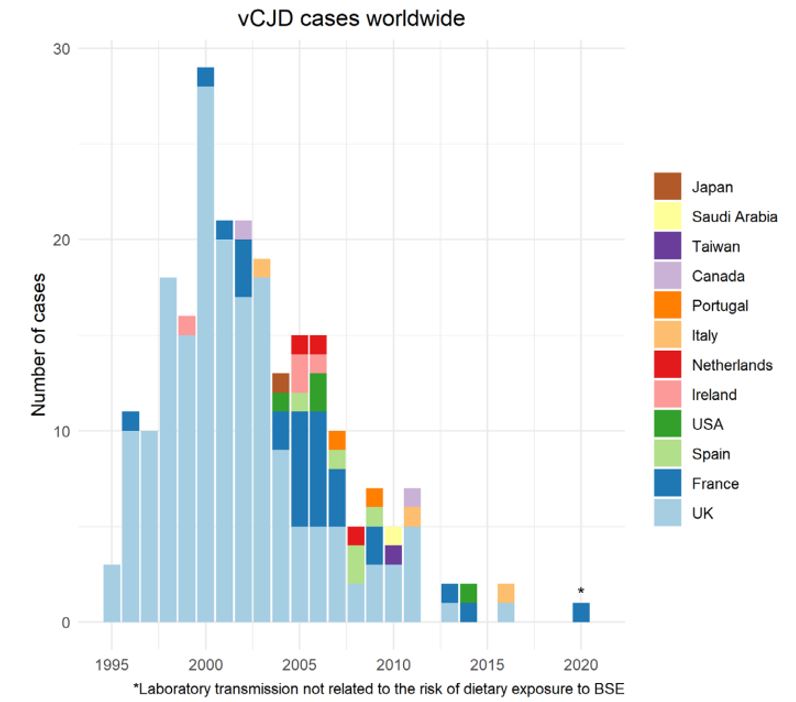
Figuur 1: Gerapporteerde gevallen van variant Creutzfeldt-Jakob (vCJD) wereldwijd tot maart 2021
Bron: Creutzfeldt-Jakob Disease International Surveillance Network en casus rapport uit Frankrijk in 2020 (Brandel 2020, McManus 2022).
Voorkomen in Nederland
CJD
De incidentie van CJD in Nederland is vergelijkbaar met de wereldwijde incidentie. De jaarlijkse incidentie in Nederland is licht toegenomen sinds 2010. Zie ook dit artikel met een overzicht van 29 jaar surveillance van prionziekten in Nederland. Tot 2009 werd gemiddeld 1,1 per miljoen inwoners per jaar gediagnosticeerd met prionziekte, in de periode 2010-2021 waren dit er gemiddeld 1,8 per miljoen per jaar (Karamujić-Čomić 2022). Wat betreft genetische CJD, zijn er van 1993-2021 in Nederland 28 patiënten met een genetische prionziekte geregistreerd, dat is 5% van het totale aantal geregistreerde prionziekten (Karamujić-Čomić 2022).
vCJD
Tot 2022 is in Nederland de diagnose variant-CJD gesteld bij drie patiënten: in 2005, 2006 en 2009. Ondanks dat de hersenobductie sinds 2022 niet meer wordt uitgevoerd in Nederland (alleen op verzoek van de nabestaanden), bestaat er nog steeds actieve surveillance voor vCJD.
Pathogenese
Bij prionziekten vindt er een conformationele verandering (andere vouwing) plaats van het normale prioneiwit PrPC naar het pathologische eiwit PrPSc. De verkeerd gevouwen prioneiwitten laten ook de nabijgelegen prioneiwitten van vorm veranderen (Prusiner 1998), deze pathogenese is uniek voor prionziekte. Door de stapeling van deze afwijkende prioneiwitten in hersenweefsel ontstaan progressieve neurodegeneratieve afwijkingen, met de dood tot gevolg. De prioneiwitten worden gecodeerd door het PRNP-gen op chromosoom 20. Genetische predispositie in dit PRNP-gen speelt een rol in zowel de kans op ontwikkelen van prionziekte als het klinisch beloop. Variatie in codon 129 is daarbij een genetische determinant, coderend voor methionine (M) en valine (V) (Parchi 2012, Parchi 1999).
CJD
Patiënten in Nederland met klinisch manifeste sporadische CJD zijn vaak homozygoot (71%) voor methionine (MM) op codon 129 van het PRNP-gen (Jansen 2012). Recent is aangetoond dat er naast codon 129 meer genetische risicovarianten zijn die een rol spelen in het ziekteproces van sporadische CJD. De genetische varianten verhogen het risico op prionziekte door de sequentie van het prioneiwit te veranderen of door een verhoogde expressie in hersenweefsel (Jones 2020).
vCJD
Ook voor vCJD is het genotype MM een risicofactor. Eerder waren er alleen homozygote MM-dragers bekend die vCJD ontwikkelden, in 2017 is er ook een MV-drager met vCJD gerapporteerd (Jansen 2012, Mok 2017). In tegenstelling tot sporadische CJD spelen bij vCJD de lymfoïde organen een rol in de pathogenese. Bij vCJD hebben prionen een voorkeurslocatie voor de tonsillen en blindedarm (Glatzel 2004, Hill 1999). Dit is relevant in relatie tot de risicogroepen voor vCJD.
Incubatieperiode
CJD
De iatrogene vorm van CJD kan een zeer lange incubatietijd hebben, tot wel 40 jaar in het geval van besmettingen uit hypofysair groeihormoon (Rudge 2015).
vCJD
De exacte incubatietijd van vCJD is niet bekend, maar betreft vaak meerdere jaren. Daarnaast kunnen geïnfecteerde individuen asymptomatisch zijn. De eerste gevallen in het Verenigd Koninkrijk vonden plaats ongeveer twaalf jaar na het hoogtepunt van de BSE-epidemie bij runderen (en blootstelling, m.a.w. consumptie van besmette vleesproducten). Hoewel de epidemie voorbij lijkt, kan het zijn dat er een reservoir van dragers is: asymptomatische aanwezigheid van prionen werd vastgesteld in weefsels van patiënten in het Verenigd Koninkrijk met niet-homozygote codon 129-genotypes (Ironside 2006, Gill 2020).
Ziekteverschijnselen
CJD
De gemiddelde leeftijd bij de start van symptomen door sporadische CJD is beschreven in de literatuur als 62 jaar (Ladogana 2005). In de database van de Landelijke Registratie Prionziekten in Nederland is de gemiddelde leeftijd bij aanvang 67,0 jaar. De uitingsvormen kunnen verschillen, mede afhankelijk van de genetische variatie op het codon 129-gen van de patiënt en van diverse andere moleculaire kenmerken van de verkeerd gevouwen prionen (Jansen 2012). Een gemeenschappelijk beloop is de snelle neuro-psychiatrische achteruitgang in een relatief kort tijdsbestek, vaak binnen een jaar, die altijd een fatale afloop heeft. In uitzonderlijke gevallen duurt het 4 jaar. Noodzakelijk voor de diagnose CJD is een snel progressieve dementie, met verschijnselen zoals gedragsveranderingen, initiatiefloosheid, vergeetachtigheid en moeite met dagelijkse handelingen. Andere kernsymptomen zijn onregelmatige spierschokken (myoklonieën), coördinatiestoornissen (ataxie), piramidale verschijnselen (verlamming, verhoogde reflexen, spasticiteit), extrapiramidale verschijnselen (stijfheid, tremoren, maskergelaat), visuele stoornissen en in het eindstadium een toestand waarin een patiënt nergens meer op reageert en niet meer beweegt of spreekt (akinetisch mutisme). Hiernaast zijn epileptische aanvallen, spraak- en slikproblemen, hallucinaties en urine-incontinentie veel voorkomende problemen (Thompson 2014).
vCJD
In tegenstelling tot de sporadische CJD treft de variantvorm meestal jongere mensen. De gemiddelde leeftijd bij het begin van symptomen is 29 jaar (16-48 jaar) (Zeidler 1997), al is er in de literatuur een grote spreiding in leeftijd beschreven: 11-74 jaar (Ironside 2017). Kenmerkend voor de variantvorm zijn vroege psychiatrische symptomen en (pijnlijke) gevoelsstoornissen, die aan het begin van de ziekte optreden. In later stadium van de ziekte kunnen ook alle andere neurologische symptomen (zoals bij sporadische CJD; cognitieve achteruitgang, myoclonus, ataxie, pyramidale- en extrapyramidale verschijnselen) erbij komen. De chronologie van de symptomen is dus belangrijk om het onderscheid met andere vormen van CJD te kunnen maken. Na twaalf tot veertien maanden overlijdt de patiënt.
Natuurlijke immuniteit
Omdat het prioneiwit een lichaamseigen eiwit is, treedt er geen immuunreactie tegen op.
Reservoir
Voor de iatrogene vorm van CJD kan de mens als reservoir worden beschouwd. Voor vCJD zijn herkauwers met BSE het meest relevante reservoir.
Transmissie
Besmettingsweg
CJD
In de normale omgang, seksueel contact, contact met normaal uitgescheiden lichaamsvloeistoffen of verpleging is er geen risico op overdracht van prionen. Een besmettingsgevaar bestaat alleen bij direct contact met specifieke weefsels of bij contact met instrumentarium dat hiermee in aanraking is geweest. Het is niet bekend vanaf welk moment voor de klinische manifestatie de risicoweefsels van een CJD-patiënt besmettelijk zijn. Een overzicht van de risico's per weefselcategorie is te vinden in de WIP-richtlijn Prionziekten en aankomende SRI-richtlijn Prionziekten. Zie ook Besmettelijkheid.
Tot nu toe is iatrogene transmissie bijna alleen opgetreden na toediening van uit kadavers gewonnen humaan groei- of gonadotropinehormoon en dura matertransplantaten (Lyodura). Zeer sporadisch ook na het gebruik van besmette elektroden direct in de hersenen, wat nu niet meer voorkomt. Deze bronnen zijn erkend als oorzaak van iatrogene besmettingen in de jaren tachtig en werden daarna wereldwijd verboden. Gezien de incubatietijd van iatrogene CJD, die tot 40 jaar kan bedragen, is het niet uitgesloten dat er nog nieuwe gevallen zullen volgen (Rudge 2015).
Ook gebruikt neurochirurgisch instrumentarium kent een theoretisch risico van iatrogene transmissie, omdat gebruikelijke sterilisatiemethoden prioneiwitten niet vernietigen. Besmetting via direct contact met hersenen en hersenvocht afkomstig van patiënten en/of overledenen is niet uitgesloten, maar er zijn tot nu toe geen gevallen bekend van besmetting op deze manier. Overdracht via bloed, bloedproducten, huidcontact, speeksel, urine, ontlasting of seksueel contact is nooit aangetoond. Bij twijfel over een risico op overdracht van prionen, dient altijd contact opgenomen te worden met de lokale infectiepreventie-afdeling van de zorginstelling.
vCJD
vCJD wordt door consumptie van met BSE besmette voedingsmiddelen overgedragen (zie Relevante transmissieroutes bij dieren). Het is onbekend of transmissie mogelijk is ten gevolge van een eenmalige hoge expositie of dat sprake moet zijn van een langdurige blootstelling.
Er zijn geen aanwijzingen dat mensen via direct contact met klinisch zieke runderen worden besmet. Het drinken van melk van klinisch zieke koeien geeft een verwaarloosbaar besmettingsrisico.
Mens-op-mensbesmetting via bloed- of plasmatransfusie is bij vijf personen vastgesteld in het Verenigd Koninkrijk (Thomas et al., 2023). Besmetting via direct en indirect contact (instrumentarium) met hersenen en hersenvocht afkomstig van patiënten en/of overledenen is niet uitgesloten. Er zijn twee gevallen gerapporteerd (in Frankrijk en Italië) van laboratoriumanalisten die mogelijk geïnfecteerd zijn geraakt via percutane blootstelling aan weefsel met vCJD erin. Echter, daarbij kon infectie via voedsel niet worden uitgesloten (Brandel et al., 2020). Overdracht via huidcontact, speeksel, urine, ontlasting of seksueel contact is niet aangetoond.
Besmettelijke periode
CJD
Besmet weefsel of besmette hormoonpreparaten blijven permanent besmettelijk.
vCJD
De besmettelijke periode verschilt per soort weefsel (zie Besmettelijkheid).
Besmettelijkheid
CJD
Het risico op besmetting is niet voor alle weefsels en vloeistoffen gelijk: er is sprake van een hoog besmettingsrisico bij direct of indirect contact met hersenen, hersenzenuwen, hypofyse, het achterste deel van het oog en ruggenmerg (ACDP TSE 2023). Prionen zijn geen levende organismen, ze kunnen daarom niet gedood worden en blijven onder diverse omstandigheden intact (ook bij normaal gebruikte desinfectiemethoden). Prionen kunnen alleen met grote moeite vernietigd worden. Het lichamelijk overschot is besmettelijk, met name de hersenen. Zie Maatregelen ten aanzien van index, contacten en bron.
vCJD
Het risico op besmetting is niet voor alle weefsels en vloeistoffen gelijk. Er bestaat een hoog besmettingsrisico bij direct of indirect contact met de volgende weefsels: appendix, dura mater, hersenen, hypofyse, lymfklieren, milt, oog, ruggenmerg, terminale ileum (plaques van Peyer) en tonsillen. Bloed van vCJD-patiënten of -dragers is in beperkte mate besmettelijk. De meeste besmettingsrisico's vinden plaats tijdens procedures in ziekenhuizen. Een overzicht van de risico's per weefselcategorie is te vinden in de WIP-richtlijn Prionziekten en aankomende SRI-richtlijn Prionziekten.
Risicogroepen
Verhoogde kans op infectie
CJD
Een potentiële risicogroep voor het ontwikkelen van CJD zijn mensen die in het verleden medische ingrepen hebben ondergaan waarbij gebruik is gemaakt van besmet instrumentarium of besmet materiaal.
Gezien de besmettingsweg, is een potentiële risicogroep het zorgpersoneel werkend aan procedures waarbij voornamelijk het centraal zenuwstelsel aangetast is/wordt. Er zijn echter geen gevallen bekend waarbij bewezen is dat CJD is opgelopen door werkzaamheden in de gezondheidszorg.
vCJD
De voornaamste risicogroep betreft personen die met BSE besmette producten hebben gegeten. Dit zou in Nederland gebeurd kunnen zijn vóór 1997. Tijdens die epidemie waren jonge mensen een risicogroep, mogelijk heeft dit te maken met dat zij meer lymfoïde weefsel in de darm hebben en vCJD-prionen een voorkeur voor lymfoïde weefsel vertonen (zie Pathogenese). Na 1997 zijn extra maatregelen genomen om te voorkomen dat met BSE besmette producten in de voedselketen zouden kunnen komen. Omdat de incubatietijd van vCJD zeer lang kan zijn, zijn ouderen (die in het verleden blootgesteld zijn) nu een potentiële risicogroep.
Behandeling
CJD
Er is geen genezing voor CJD mogelijk. Trials met medicatie worden bemoeilijkt door de zeldzaamheid van de ziekte en het feit dat de diagnose pas gesteld wordt als de pathologische processen al vergevorderd zijn (Geschwind 2008). De behandeling van een CJD-patiënt is gericht op symptoombestrijding en comfort.
vCJD
Er is vooralsnog geen behandeling die tot genezing leidt. Experimenten met intrathecaal pentosanpolysulfaat hebben wisselende resultaten laten zien in het vertragen van het ziekteproces.
Diagnostiek
Microbiologische diagnostiek
Geen.
Niet-microbiologische diagnostiek
Voor algemene vragen omtrent diagnostiek van CJD kan contact worden opgenomen met de Landelijke Registratie Prionziekten, via de afdeling Epidemiologie van het Erasmus (Medisch Centrum) (010-7043391; info.cjd@erasmusmc.nl). De diagnostiek om tot een (waarschijnlijkheids)diagnose te komen, wordt uitgevoerd in verschillende laboratoria en klinieken, zie hieronder.
CJD
Indien na anamnese en lichamelijk onderzoek een klinische verdenking op CJD bestaat moet aanvullend onderzoek worden ingezet om een waarschijnlijkheidsdiagnose te stellen. Volgens de diagnostische criteria kan de diagnose al worden gesteld op basis van progressieve dementie in combinatie met een positieve prion Real-Time Quaking-Induced Conversion (RT-QuIC) test. Een negatieve RT-QuIC-testuitslag sluit CJD niet helemaal uit en bij blijvende twijfel moet de test worden herhaald.
Gezien de uitslag enige tijd duurt en er een differentiaal diagnose bestaat, wordt ook een MRI-scan van de hersenen gemaakt. Specifiek afwijkend bij CJD is een hoog signaal in de basale kernen en corticale gebieden (op 'fluid-attenuated inversion recovery' (FLAIR)-opnames en/of diffusiegewogen imaging (DWI)). MRI-afwijkingen in de basale kernen en/of de cortex worden bij ca. 83% van de gevallen gevonden, maar zijn nog niet voldoende voor diagnosestelling (Meissner 2009, Zerr 2009).
Overleg bij twijfel met experts van de Landelijke Registratie Prionziekten: 010-7043391 of de gespecialiseerde dementie-centra (Alzheimercentrum AUMC en ErasmusMC).
Hersenvochtonderzoek
Tot 2022 werd pathologisch onderzoek van hersenweefsel, verkregen bij obductie, als de gouden standaard beschouwd voor de diagnose van CJD. Bij histopathologisch onderzoek worden kenmerkende veranderingen gezien, waaronder spongiforme degeneratie, neuronverlies en astrocytaire gliose (Budka 2003, Parchi 1999). Hersenobductie is in Nederland geen standaardonderdeel meer van de diagnostiek voor CJD (alleen als de nabestaanden dit willen, op eigen kosten). Tegenwoordig wordt de RT-QuIC-test ingezet voor bevestiging van de diagnose (Green 2019). Dit is een zogenaamde eiwit amplificatietest, die uitgevoerd wordt in het hersenvocht, waarbij het pathologische prioneiwit (PrPSc) in hersenvocht wordt gedetecteerd. De test is gebaseerd op dit principe: als er verkeerd gevouwen PrPSc eiwit aanwezig is in hersenvocht, kan dit het exogeen toegevoegd, correct gevouwen, PrPC eiwit omzetten tot PrPSc eiwit. De toegenomen hoeveelheid van dit verkeerd gevouwen PrPSc eiwit wordt real-time gevolgd met behulp van fluorescerende kleurstoffen. De RT-QuIC-test heeft een sensitiviteit van 91-92% en een specificiteit van 98-100% (Green 2019, (European Centre for Disease Prevention and Control) 2023). Echter, indien de test te vroeg in het ziekteproces wordt ingezet, kan deze nog negatief zijn. Bij blijvende klinische verdenking op CJD kan daarom overwogen worden de RT-QuIC-test te herhalen. Twee laboratoria in Nederland hebben de specialisatie de RT-QuIC-test uit te voeren: het Radboud (Universitair Medisch Centrum Utrecht) te Nijmegen (Neurochemisch Laboratorium; zie hiervoor: Translationeel Metabool Laboratorium - Radboudumc) en het UMC Utrecht (Prionlab). Meer informatie over de RT-QuIC-test is hier beschikbaar of via de volgende link: eLabgids (getincontrol.eu) (Radboudumc).
Dan is er nog het 14-3-3 eiwit, gemeten in het hersenvocht. Dit is een aspecifieke marker voor snel progressief verval van hersenweefsel, die bij veel snel verlopende hersenziekten in het hersenvocht wordt gevonden. De specificiteit voor het uitsluiten van CJD in klinische studies ligt doorgaans rond de 90% en de sensitiviteit voor het aantonen van CJD is rond de 75% (Stoeck 2012), maar in de praktijk kunnen nog lagere waarden worden gevonden (Geschwind 2003). Mede hierdoor fungeert het 14-3-3 eiwit enkel als een aanvullende marker, naast klinische beoordelingen en andere diagnostische onderzoeken.
Een andere marker die in het hersenvocht wordt aangetroffen is een verhoogd totaal tau-eiwit bij normaal gefosforyleerd tau-eiwit (ofwel een verhoogde totaal-tau/gefosforyleerde tau-ratio), dit wijst op versneld verval van axonen (Blennow 2005). Er wordt bij CJD vrijwel altijd een sterk verhoogde totaal-tau/gefosforyleerd tau-ratio aangetroffen; een normale ratio sluit met grote mate van zekerheid de diagnose CJD uit. Deze analyses kunnen dus zeer behulpzaam zijn bij een lage klinische verdenking op CJD om de diagnose uit te sluiten. Andere eiwitten die bij CJD in (zeer) hoge concentraties in het hersenvocht worden aangetroffen zijn S-100b, neuronspecifiek enolase (NSE) en neurofilament light chain (NFL).
Het is daarnaast belangrijk alert te zijn op behandelbare mimics van CJD, zoals auto-immuun encefalitis, al dan niet op paraneoplastische basis (Geschwind 2008). Screening op deze anti-neuronale antistoffen, inclusief anti-voltage-gated kaliumkanalen (VGKC) en de anti-N-methyl-D-aspartaat (NMDA)-antistoffen en anti-schildklier antistoffen, moet daarom altijd worden overwogen. Ook centraal zenuwstelselinfecties dienen uitgesloten te worden (Simon & Peter 2017). Onderzoek naar paraneoplastische/anti-neuronale antilichamen vindt plaats in het Radboudumc (Neurochemie laboratorium) of in het Erasmus MC Rotterdam (afdeling Immunologie).
Voor vragen over het hersenvochtonderzoek dan wel het immunologisch onderzoek kan overlegd worden met de betreffende laboratoria.
Elektro-encefalografie (EEG)
Bij een deel van de patiënten kan het EEG een karakteristiek patroon laten zien, de zogenaamde trifasische golven. Deze golven worden vaak later in het ziekteproces waargenomen en komen bij 50-75% van de gevallen voor, afhankelijk van het subtype CJD (Wieser 2006).
Bij positieve familieanamnese en/of specifieke klinische beelden die duiden op een genetische vorm van CJD is genetisch onderzoek en verwijzing naar een klinisch geneticus geïndiceerd.
vCJD
In verband met een hoog risico op transmissie van prionen, moet een verdenking op vCJD binnen 24 uur gemeld worden bij de Landelijke Registratie Prionziekten, via de afdeling Epidemiologie van het Erasmus MC (010-7043391; info.cjd@erasmusmc.nl). Hier kunnen tevens vragen gesteld worden omtrent de diagnostiek. Zie ook Maatregelen.
Patiënten met de variantvorm presenteren zich meestal anders dan patiënten met de sporadische vorm. Bij de variantvorm staan aan het begin van de ziekte psychiatrische symptomen (depressie, angst, emotionele terugtrekking) op de voorgrond. Andere neurologische symptomen ontwikkelen zich later. Daarnaast kan er sprake zijn van persisterende pijnlijke sensorische stoornissen (CDC 2021, ECDC 2017). Alleen post mortem neuropathologisch onderzoek kan de diagnose vCJD compleet bevestigen. Histopathologisch vertoont de ziekte kenmerken van spongiforme veranderingen en uitgebreide afzettingen van prioneiwitten, met opvallende floride plaques in zowel het cerebrum als het cerebellum. Deze bevindingen zijn duidelijk verschillend van andere vormen van CJD (Ironside & Head 2004). Antemortem laat een EEG in de vroege fase niet de typische verschijnselen van sporadische CJD zien (Will 2000) en ook de RT-QuIC-test valt bij vCJD negatief uit (Green 2019, Orrú 2015). Op een MRI-scan kan echter wel een bilateraal symmetrisch verhoogd signaal in het pulvinar worden waargenomen. Een tonsilbiopt kan aanvullende aanwijzingen bieden voor de aanwezigheid van vCJD, maar dit wordt in de Nederlandse praktijk weinig meer toegepast. Een tonsilbiopt is vooral nuttig bij klinisch verdachte patiënten met een negatieve MRI. De diagnostiek van hersenvocht heeft een veel lagere gevoeligheid dan bij de klassieke/sporadische vorm van CJD. Uitslag van de RT-QuIC-test kan vCJD niet bevestigen of uitsluiten, deze test is niet ontwikkeld voor vCJD.
Preventie
Immunisatie
Vaccinatie
Geen.
Algemene preventieve maatregelen
CJD
Het treffen van algemene preventieve maatregelen ter voorkoming van iatrogene transmissie van CJD is met name van toepassing binnen het ziekenhuis. In ziekenhuizen en klinieken waar met risicomateriaal wordt gewerkt moeten algemene preventieve maatregelen worden genomen om te voorkomen dat potentieel aanwezige prioneiwitten op instrumentarium of in liquor in direct contact komen met risicoweefsel van patiënten en medewerkers. Zie hiervoor verder de WIP-richtlijn Prionziekten en aankomende SRI-richtlijn Prionziekten, die beschrijft wat te doen met (mogelijk) gecontamineerd instrumentarium.
vCJD
Preventie bestaat uit het voorkomen van BSE en voorkomen dat met BSE besmette producten terechtkomen in de voedselketen. Dit is bereikt door dierlijke producten volledig te weren uit voer voor herkauwers, waardoor besmettelijke BSE grotendeels geëlimineerd is (m.u.v. de atypische 'ouderdoms-BSE', zie ook Epidemiologie).
Er is daarnaast ook nog een klein risico op overdracht van vCJD via bloedtransfusie. Daarom wordt in Nederland leukodepletie toegepast: verwijdering van witte bloedcellen uit donorbloed (leukodepletie) vermindert de infectiviteit van bloed (Gregori 2004, van Aken 2001). Er is geen methode om betrouwbaar de aanwezigheid van prionen in donorbloed aan te tonen (McManus 2022).
Reiniging, desinfectie en sterilisatie
Bij verdenking op vCJD of wanneer vCJD is vastgesteld bij een patiënt moeten bij medische ingrepen aanvullende maatregelen worden getroffen. Welke maatregelen dat zijn, is afhankelijk van het besmettingsrisico (welke weefsels en/of vloeistoffen) en in welke mate de diagnose vCJD vaststaat. Voor de juiste werkwijze wordt verwezen naar de WIP-richtlijn Prionziekten en aankomende SRI-richtlijn Prionziekten.
De standaardmethoden voor reiniging, desinfectie en steriliseren zijn niet toereikend voor het inactiveren van prionen. Instrumenten die gebruikt zijn bij weefsels die aangemerkt zijn als een hoog of middelhoog risico, dienen na gebruik worden weggegooid of enkel hergebruikt te worden bij dezelfde patiënt.
Maatregelen
Meldingsplicht
CJD is een meldingsplichtige ziekte, groep C volgens de Wet publieke gezondheid. De meldingsprocedure wijkt af van die van andere meldingsplichtige aandoeningen. Zie voor de rollen en taakverdeling de bijlage Extramurale procedures bij melding van de ziekte van Creutzfeldt-Jakob.
CJD
De behandelend arts en/of het laboratorium dient zowel een waarschijnlijke als vastgestelde diagnose van CJD binnen één werkdag te melden aan de Landelijke Registratie Prionziekten van het Erasmus MC: 010-7043391.
Toelichting: In de praktijk worden er in Nederland geen obducties meer gedaan bij verdenkingen van (v)CDJ. Hierdoor komt het zelden tot nooit tot een bevestigde diagnose van CJD maar meestal tot een (zeer) waarschijnlijke diagnose (zie ook de criteria voor de melding en paragraaf Diagnostiek), vandaar dat de behandelend arts de waarschijnlijke diagnose dient te melden.
De Landelijke Registratie Prionziekten verifieert de diagnose en geeft binnen twee werkdagen de melding door aan de afdeling infectieziektebestrijding van de (Gemeentelijke gezondheidsdienst) (de GGD in de regio waar de patiënt woont). De GGD registreert de melding in Osiris (binnen één week). De Landelijke Registratie Prionziekten neemt vervolgens contact op met de patiënt en diens familie via online consultaties en bespreekt de optie tot deelname aan wetenschappelijk onderzoek (en het belang daarvan) in de vorm van een vragenlijst en bloedafname.
Meldingscriteria CJD:
Let op: Zowel waarschijnlijke CJD als vastgestelde CJD moet worden gemeld.
Vastgestelde CJD
Neuropathologische bevestiging (door hersenobductie of biopsie).
Waarschijnlijke CJD
Ziektebeeld gekenmerkt door progressieve dementie
in combinatie met
- twee of meer van de volgende vier verschijnselen:
- myoclonus
- visuele of cerebellaire stoornissen
- piramidale of extrapiramidale verschijnselen
akinetisch mutisme
in combinatie met;
- karakteristiek EEG
en/of
- hyperintense signaal van de basale kernen op Flair of DWI-serie op MRI
en/of
- positieve 14-3-3 liquortest en duur < 2 jaar.OF
- een positieve RT-QuIC test.
Zie voor toelichting van de diagnostische stappen de paragraaf Diagnostiek.
vCJD
De behandelend arts en/of het laboratorium dient zowel een waarschijnlijke als vastgestelde diagnose van vCJD binnen 24 uur te melden aan de Landelijke Registratie Prionziekten van het Erasmus MC: 010-7043391.
Toelichting: In de praktijk worden er in Nederland geen obducties meer gedaan bij verdenkingen van (v)CDJ, hierdoor komt het zelden tot nooit tot een bevestigde diagnose van (v)CJD, maar meestal tot een (zeer) waarschijnlijke diagnose (zie ook de criteria voor de melding en de paragraaf Diagnostiek), vandaar dat de behandelend arts de waarschijnlijke diagnose dient te melden.
De Landelijke Registratie Prionziekten neemt na verificatie van de verdenking, binnen 24 uur telefonisch contact op met de (Landelijke Coördinatie Infectieziektebestrijding) en GGD. LCI en GGD hebben vervolgens binnen 24 uur contact over te nemen maatregelen. De GGD registreert elektronisch de melding in OSIRIS (binnen 24 uur).
Meldingscriteria vCJD
Let op: Zowel waarschijnlijke vCJD als vastgestelde vCJD moet worden gemeld.
Vastgestelde vCJD
Progressieve neuropsychiatrische stoornis met neuropathologische bevestiging van vCJD (spongiose en uitgebreide PrP-deposities in cerebrum en cerebellum met floride plaques).
Waarschijnlijke vCJD
Ziektebeeld gekenmerkt door:
- progressieve neuropsychiatrische stoornis
- duur van de ziekte > 6 maanden
- waarbij routineonderzoek niet duidt op een alternatieve diagnose
- er geen voorgeschiedenis is van potentieel iatrogene blootstelling
- er geen bewijs bestaat voor een familiaire vorm van de overdraagbare spongiforme encefalopathieën
in combinatie met
vier van de vijf volgende verschijnselen:
- vroege psychiatrische symptomen
- aanhoudende pijnlijke sensibiliteitsstoornissen
- ataxie
myoclonus
of
chorea
of
dystonie
- dementie
in combinatie met:
- een EEG dat in een vroeg stadium niet de typische verschijnselen van sporadische CJD vertoont (in een laat stadium van vCJD kunnen soms trifasische periodieke complexen worden gezien)
in combinatie met:
een bilateraal symmetrisch verhoogd signaal in het pulvinar op de MRI-scan
OF
Een ziektebeeld gekenmerkt door:
- progressieve neuropsychiatrische stoornis
- duur van de ziekte > 6 maanden
- waarbij routine-onderzoek niet duidt op een alternatieve diagnose
- er geen voorgeschiedenis is van potentieel iatrogene blootstelling
- er geen bewijs bestaat voor een familiaire vorm van de overdraagbare spongiforme encefalopathieën
in combinatie met
- positief tonsil biopt.
Inschakelen van andere instanties
Zie voor de rollen en taakverdeling de bijlage Extramurale procedures bij melding van de ziekte van Creutzfeldt-Jakob.
Bron- en contactonderzoek
Bronopsporing
CJD
Geen. Eventueel kan een onderzoek worden ingesteld als er zich een bijzonder aantal casus in een bepaalde regio voordoen in korte tijd.
vCJD
Bij vCJD wordt nagegaan hoe de patiënt kan zijn besmet. Zie voor de rollen en taakverdeling de bijlage Extramurale procedures bij melding van de ziekte van Creutzfeldt-Jakob.
Contactonderzoek
CJD
Het ziekenhuis of de instelling waarin de patiënt ligt of is overleden of een medische ingreep heeft ondergaan, is verantwoordelijk voor het beoordelen van het besmettingsrisico en eventueel benodigde inventarisatie van personen (patiënten en medewerkers) die mogelijk risico hebben gelopen op besmetting in het ziekenhuis. Zie de WIP-richtlijn Prionziekten en aankomende SRI-richtlijn Prionziekten.
vCJD
Het ziekenhuis waar de patiënt heeft gelegen, is overleden of een medische ingreep heeft ondergaan, is verantwoordelijk voor het beoordelen van het besmettingsrisico en eventueel benodigde inventarisatie van personen (patiënten en medewerkers) die mogelijk risico hebben gelopen op besmetting in het ziekenhuis. Zie de WIP-richtlijn Prionziekten en aankomende SRI-richtlijn Prionziekten. De GGD heeft alleen een rol in de inventarisatie van ingrepen en mogelijke risico's buiten het ziekenhuis of de zorginstelling, in de praktijk komt dit heel weinig voor. De IGJ zorgt, indien van toepassing, voor het uit de roulatie nemen van eventueel resterende bloedproducten en andere gedoneerde weefsels van de patiënt.
Maatregelen ten aanzien van index, contacten en bron
CJD
Het ziekenhuis of de zorginstelling onderzoekt de besmettingsrisico’s en zal daarnaast informatie aan de direct betrokkenen verstrekken. De belangrijkste vragen zullen zorg voor de patiënt en preventie van mogelijk secundaire transmissie betreffen. De Landelijke Registratie Prionziekten (Afdeling Epidemiologie, Erasmus MC) is beschikbaar voor overleg voor artsen, GGD en familieleden. De GGD kan zo nodig vragen van het algemene publiek of de pers beantwoorden (in samenwerking met het ziekenhuis).
Indien er een risico is op besmetting van anderen via instrumentarium dat in contact is geweest met hoog-risicoweefsel van een patiënt, neemt het ziekenhuis indien mogelijk het instrumentarium direct in quarantaine bij de eerste verdenking, of vernietigt het bij een zekere diagnose of overlijden zonder obductie. Het ziekenhuis verricht een risico-inschatting en zo nodig worden patiënten die mogelijk zijn blootgesteld geïdentificeerd. De GGD is hierbij betrokken. De LCI en de Landelijke Registratie Prionziekten kunnen worden geconsulteerd om te beoordelen of er verdere stappen/maatregelen nodig zijn.
Het lichamelijk overschot is potentieel besmettelijk. Er is geen besmettingsrisico bij opbaren en begraven of cremeren van het lichaam (zie Besmettelijkheid). Balseming dient vermeden te worden en bij uitzondering alleen verricht door speciaal geschoold personeel. Geadviseerd wordt aan academische centra om het lichaam van een patiënt met vermoedelijk CJD niet als anatomisch specimen te accepteren.
vCJD
Het ziekenhuis verstrekt informatie aan direct betrokkenen. De GGD beantwoordt in samenspraak met het ziekenhuis eventuele vragen van lokale media en het algemeen publiek. De landelijke pers wordt verwezen naar de persvoorlichting van het (Rijksinstituut voor Volksgezondheid en Milieu). Ook de Landelijke Registratie Prionziekten is beschikbaar voor overleg voor GGD, ziekenhuizen en direct betrokkenen.
Wanneer de GGD de melding van een waarschijnlijke of vastgestelde vCJD krijgt, overlegt zij met de LCI over de noodzakelijke gegevensverzameling ten aanzien van de patiënt. In tegenstelling tot bij de sporadische vorm van de ziekte, kan obductie zo nodig juridisch worden afgedwongen in het belang van de publieke gezondheid; tot nu toe is dit in Nederland niet nodig geweest. Zie verder de bijlage Extramurale procedures bij melding van de ziekte Creutzfeldt-Jakob.
Postexpositieprofylaxe
Geen.
Wering
Geen.
Literatuur
- ACDP-TSE. (2023). Guidance Minimise transmission risk of CJD and (variant Creutzfeldt-Jakob Disease) in healthcare settings. Retrieved June 2 from https://www.gov.uk/government/publications/guidance-from-the-acdp-tse-r….
- Aguzzi, A., Montrasio, F., & Kaeser, P. S. (2001). Prions: health scare and biological challenge. Nature Reviews Molecular Cell Biology, 2(2), 118-126. https://doi.org/10.1038/35052063.
- Babelhadj, B., Di Bari, M. A., Pirisinu, L., Chiappini, B., Gaouar, S. B. S., Riccardi, G., Marcon, S., Agrimi, U., Nonno, R., & Vaccari, G. (2018). Prion Disease in Dromedary Camels, Algeria. Emerg Infect Dis, 24(6), 1029-1036. https://doi.org/10.3201/eid2406.172007.
- Blennow, K., Johansson, A., & Zetterberg, H. (2005). Diagnostic value of 14-3-3beta immunoblot and T-tau/P-tau ratio in clinically suspected Creutzfeldt-Jakob disease. Int J Mol Med, 16(6), 1147-1149.
- Brandel, J.-P., Vlaicu, M. B., Culeux, A., Belondrade, M., Bougard, D., Grznarova, K., Denouel, A., Plu, I., Bouaziz-Amar, E., Seilhean, D., Levasseur, M., & Haïk, S. (2020). Variant Creutzfeldt–Jakob Disease Diagnosed 7.5 Years after Occupational Exposure. New England Journal of Medicine, 383(1), 83-85. https://doi.org/10.1056/NEJMc2000687.
- Brandel, J. P., Heath, C. A., Head, M. W., Levavasseur, E., Knight, R., Laplanche, J. L., Langeveld, J. P., Ironside, J. W., Hauw, J. J., Mackenzie, J., Alpérovitch, A., Will, R. G., & Haïk, S. (2009). Variant Creutzfeldt-Jakob disease in France and the United Kingdom: Evidence for the same agent strain. Ann Neurol, 65(3), 249-256. https://doi.org/10.1002/ana.21583.
- Budka, H. (2003). Neuropathology of prion diseases. British Medical Bulletin, 66(1), 121-130. https://doi.org/10.1093/bmb/66.1.121.
- Carlson, G. A., & Prusiner, S. B. (2021). How an Infection of Sheep Revealed Prion Mechanisms in Alzheimer's Disease and Other Neurodegenerative Disorders. Int J Mol Sci, 22(9). https://doi.org/10.3390/ijms22094861.
- CDC. (2021, 18-10-2021). Variant Creutzfeldt-Jakob Disease (vCJD). Retrieved 30-06-2023 from https://www.cdc.gov/prions/vcjd/diagnostic-criteria.html.
- Corbett, G. T., Wang, Z., Hong, W., Colom-Cadena, M., Rose, J., Liao, M., Asfaw, A., Hall, T. C., Ding, L., DeSousa, A., Frosch, M. P., Collinge, J., Harris, D. A., Perkinton, M. S., Spires-Jones, T. L., Young-Pearse, T. L., Billinton, A., & Walsh, D. M. (2020). PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol, 139(3), 503-526. https://doi.org/10.1007/s00401-019-02114-9.
- (European Centre for Disease Prevention and Control). (2017). Facts about variant Creutzfeldt-Jakob disease. Retrieved June 2023 from https://www.ecdc.europa.eu/en/vcjd/facts.
- ECDC. (2023). CJD-SOP-for-RT-QuIC-final-feb-2020.pdf. Retrieved August 2023 from https://www.ecdc.europa.eu/sites/default/files/documents/CJD-SOP-for-RT….
- EFSA. (2022). The European Union summary report on surveillance for the presence of transmissible spongiform encephalopathies (TSE) in 2021. EFSA Journal, 20(11), e07655. https://doi.org/https://doi.org/10.2903/j.efsa.2022.765.
- (Europese Unie). (2023). TSE/BSE explained. Retrieved May 2023 from https://food.ec.europa.eu/safety/biological-safety/food-borne-diseases-….
- Geschwind, M. D., Martindale, J., Miller, D., DeArmond, S. J., Uyehara-Lock, J., Gaskin, D., Kramer, J. H., Barbaro, N. M., & Miller, B. L. (2003). Challenging the Clinical Utility of the 14-3-3 Protein for the Diagnosis of Sporadic Creutzfeldt-Jakob Disease. Archives of Neurology, 60(6), 813-816. https://doi.org/10.1001/archneur.60.6.813.
- Geschwind, M. D., Tan, K. M., Lennon, V. A., Barajas, R. F., Jr., Haman, A., Klein, C. J., Josephson, S. A., & Pittock, S. J. (2008). Voltage-Gated Potassium Channel Autoimmunity Mimicking Creutzfeldt-Jakob Disease. Archives of Neurology, 65(10), 1341-1346. https://doi.org/10.1001/archneur.65.10.1341.
- Gill, O. N., Spencer, Y., Richard-Loendt, A., Kelly, C., Brown, D., Sinka, K., Andrews, N., Dabaghian, R., Simmons, M., Edwards, P., Bellerby, P., Everest, D. J., McCall, M., McCardle, L. M., Linehan, J., Mead, S., Hilton, D. A., Ironside, J. W., & Brandner, S. (2020). Prevalence in Britain of abnormal prion protein in human appendices before and after exposure to the cattle BSE epizootic. Acta Neuropathol, 139(6), 965-976. https://doi.org/10.1007/s00401-020-02153-7.
- Glatzel, M., Giger, O., Seeger, H., & Aguzzi, A. (2004). Variant Creutzfeldt-jakob disease: between lymphoid organs and brain. Trends Microbiol, 12(2), 51-53. https://doi.org/10.1016/j.tim.2003.12.001.
- Green, A.J.E. RT-QuIC: a new test for sporadic CJD. Practical Neurology 2019;19:49-55. https://doi.org/10.1136/practneurol-2018-00193.
- Greenlee, J. J., & Greenlee, M. H. (2015). The transmissible spongiform encephalopathies of livestock. Ilar j, 56(1), 7-25. https://doi.org/10.1093/ilar/ilv008.
- Gregori, L., McCombie, N., Palmer, D., Birch, P., Sowemimo-Coker, S. O., Giulivi, A., & Rohwer, R. G. (2004). Effectiveness of leucoreduction for removal of infectivity of transmissible spongiform encephalopathies from blood. Lancet, 364(9433), 529-531. https://doi.org/10.1016/s0140-6736(04)16812-8.
- Hewitt, P. E., Llewelyn, C. A., Mackenzie, J., & Will, R. G. (2006). Creutzfeldt-Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang, 91(3), 221-230. https://doi.org/10.1111/j.1423-0410.2006.00833.x.
- Hill, A. F., Butterworth, R. J., Joiner, S., Jackson, G., Rossor, M. N., Thomas, D. J., Frosh, A., Tolley, N., Bell, J. E., Spencer, M., King, A., Al-Sarraj, S., Ironside, J. W., Lantos, P. L., & Collinge, J. (1999). Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet, 353(9148), 183-189. https://doi.org/10.1016/s0140-6736(98)12075-5.
- Ironside, J. W., & Head, M. W. (2004). Neuropathology and molecular biology of variant Creutzfeldt-Jakob disease. Curr Top Microbiol Immunol, 284, 133-159.
- Ironside, J. W., Ritchie, D. L., & Head, M. W. (2017). Prion diseases. Handb Clin Neurol, 145, 393-403. https://doi.org/10.1016/b978-0-12-802395-2.00028-6.
- Jansen, C., Parchi, P., Capellari, S., Ibrahim-Verbaas, C. A., Schuur, M., Strammiello, R., Corrado, P., Bishop, M. T., van Gool, W. A., Verbeek, M. M., Baas, F., van Saane, W., Spliet, W. G., Jansen, G. H., van Duijn, C. M., & Rozemuller, A. J. (2012). Human prion diseases in the Netherlands (1998-2009): clinical, genetic and molecular aspects. PLoS One, 7(4), e36333. https://doi.org/10.1371/journal.pone.0036333.
- Jones, E., Hummerich, H., Viré, E., Uphill, J., Dimitriadis, A., Speedy, H., Campbell, T., Norsworthy, P., Quinn, L., Whitfield, J., Linehan, J., Jaunmuktane, Z., Brandner, S., Jat, P., Nihat, A., How Mok, T., Ahmed, P., Collins, S., Stehmann, C., . . . Mead, S. (2020). Identification of novel risk loci and causal insights for sporadic Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol, 19(10), 840-848. https://doi.org/10.1016/s1474-4422(20)30273-8.
- Karamujić-Čomić, H., Rozemuller, A. J. M., Verbeek, M. M., Lemstra, A. W., Ikram, M. A., & van Duijn, C. M. (2022). [Prion diseases in The Netherlands: twenty-nine years of surveillance]. Ned Tijdschr Geneeskd, 166. (Prionziekten in Nederland).
- Kovač, V., & Čurin Šerbec, V. (2022). Prion Protein: The Molecule of Many Forms and Faces. Int J Mol Sci, 23(3). https://doi.org/10.3390/ijms23031232https://doi.org/10.3390/ijms23031232.
- Ladogana, A., Puopolo, M., Croes, E. A., Budka, H., Jarius, C., Collins, S., Klug, G. M., Sutcliffe, T., Giulivi, A., Alperovitch, A., Delasnerie-Laupretre, N., Brandel, J. P., Poser, S., Kretzschmar, H., Rietveld, I., Mitrova, E., Cuesta Jde, P., Martinez-Martin, P., Glatzel, M., . . . Zerr, I. (2005a). Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology, 64(9), 1586-1591. https://doi.org/10.1212/01.Wnl.0000160117.56690.B2.
- Ladogana, A., Puopolo, M., Poleggi, A., Almonti, S., Mellina, V., Equestre, M., & Pocchiari, M. (2005b). High incidence of genetic human transmissible spongiform encephalopathies in Italy. Neurology, 64(9), 1592-1597. https://doi.org/10.1212/01.Wnl.0000160118.26865.11.
- Laurén, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., & Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature, 457(7233), 1128-1132. https://doi.org/10.1038/nature07761.
- Matamoros-Angles, A., Mohammadi, B., Song, F., Shafiq, M., Brenna, S., Puig, B., Glatzel, M., & Altmeppen, H. C. (2023). Inducing prion protein shedding as a neuroprotective and regenerative approach in pathological conditions of the brain: from theory to facts. Neural Regeneration Research, 18(9), 1869-1875. https://pubmed.ncbi.nlm.nih.gov/36926701.
- McManus, H., Seed, C. R., Hoad, V. C., Kiely, P., Kaldor, J. M., Styles, C. E., Yang, H., Law, M., & Gosbell, I. B. (2022). Risk of variant Creutzfeldt-Jakob disease transmission by blood transfusion in Australia. Vox Sang, 117(8), 1016-1026. https://doi.org/10.1111/vox.13290.
- Meissner, B., Kallenberg, K., Sanchez-Juan, P., Collie, D., Summers, D. M., Almonti, S., Collins, S. J., Smith, P., Cras, P., Jansen, G. H., Brandel, J. P., Coulthart, M. B., Roberts, H., Van Everbroeck, B., Galanaud, D., Mellina, V., Will, R. G., & Zerr, I. (2009). MRI lesion profiles in sporadic Creutzfeldt-Jakob disease. Neurology, 72(23), 1994-2001.
- Mok, T., Jaunmuktane, Z., Joiner, S., Campbell, T., Morgan, C., Wakerley, B., Golestani, F., Rudge, P., Mead, S., Jäger, H. R., Wadsworth, J. D., Brandner, S., & Collinge, J. (2017). Variant Creutzfeldt-Jakob Disease in a Patient with Heterozygosity at PRNP Codon 129. N Engl J Med, 376(3), 292-294. https://doi.org/10.1056/NEJMc1610003.
- Orrú, C. D., Groveman, B. R., Raymond, L. D., Hughson, A. G., Nonno, R., Zou, W., Ghetti, B., Gambetti, P., & Caughey, B. (2015). Bank Vole Prion Protein As an Apparently Universal Substrate for RT-QuIC-Based Detection and Discrimination of Prion Strains. PLoS Pathog, 11(6), e1004983.
- Parchi, P., de Boni, L., Saverioni, D., Cohen, M. L., Ferrer, I., Gambetti, P., Gelpi, E., Giaccone, G., Hauw, J. J., Höftberger, R., Ironside, J. W., Jansen, C., Kovacs, G. G., Rozemuller, A., Seilhean, D., Tagliavini, F., Giese, A., & Kretzschmar, H. A. (2012). Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol, 124(4), 517-529. https://doi.org/10.1007/s00401-012-1002-8.
- Parchi, P., Giese, A., Capellari, S., Brown, P., Schulz-Schaeffer, W., Windl, O., Zerr, I., Budka, H., Kopp, N., Piccardo, P., Poser, S., Rojiani, A., Streichemberger, N., Julien, J., Vital, C., Ghetti, B., Gambetti, P., & Kretzschmar, H. (1999). Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol, 46(2), 224-233.
- Prusiner, S. B. (1998). Prions. Proc Natl Acad Sci U S A, 95(23), 13363-13383. https://doi.org/10.1073/pnas.95.23.13363.
- Rhoads, D. D., Wrona, A., Foutz, A., Blevins, J., Glisic, K., Person, M., Maddox, R. A., Belay, E. D., Schonberger, L. B., Tatsuoka, C., Cohen, M. L., & Appleby, B. S. (2020). Diagnosis of prion diseases by RT-QuIC results in improved surveillance. Neurology, 95(8), e1017-e1026. https://doi.org/10.1212/wnl.0000000000010086.
- Rudge, P., Jaunmuktane, Z., Adlard, P., Bjurstrom, N., Caine, D., Lowe, J., Norsworthy, P., Hummerich, H., Druyeh, R., Wadsworth, J. D., Brandner, S., Hyare, H., Mead, S., & Collinge, J. (2015). Iatrogenic CJD due to pituitary-derived growth hormone with genetically determined incubation times of up to 40 years. Brain, 138(Pt 11), 3386-3399. https://doi.org/10.1093/brain/awv235.
- Simon, M., & Peter, R. (2017). CJD mimics and chameleons. Practical Neurology, 17(2), 113-121. https://doi.org/10.1136/practneurol-2016-001571.
- Spiropoulos, J., Lockey, R., Sallis, R. E., Terry, L. A., Thorne, L., Holder, T. M., Beck, K. E., & Simmons, M. M. (2011). Isolation of prion with BSE properties from farmed goat. Emerg Infect Dis, 17(12), 2253-2261. https://doi.org/10.3201/eid1712.110333.
- Stoeck, K., Sanchez-Juan, P., Gawinecka, J., Green, A., Ladogana, A., Pocchiari, M., Sanchez-Valle, R., Mitrova, E., Sklaviadis, T., Kulczycki, J., Slivarichova, D., Saiz, A., Calero, M., Knight, R., Aguzzi, A., Laplanche, J. L., Peoc'h, K., Schelzke, G., Karch, A., . . . Zerr, I. (2012). Cerebrospinal fluid biomarker supported diagnosis of Creutzfeldt-Jakob disease and rapid dementias: a longitudinal multicentre study over 10 years. Brain, 135(Pt 10), 3051-3061.
- Thomas, S., Roberts, B., Domanović, D., Kramer, K., Klochkov, D., Sivasubramaniyam, S., Miloslavich, D., Plançon, J. P., Rossi, F., Misztela, D., Kirkpatrick, L., Miflin, G., Birchall, J., McLintock, L., & Knight, R. (2023). Safety profile of plasma for fractionation donated in the United Kingdom, with respect to variant Creutzfeldt-Jakob disease. Vox Sang, 118(5), 345-353. https://doi.org/10.1111/vox.13416.
- Thompson, A., MacKay, A., Rudge, P., Lukic, A., Porter, M. C., Lowe, J., Collinge, J., & Mead, S. (2014). Behavioral and psychiatric symptoms in prion disease. Am J Psychiatry, 171(3), 265-274. https://doi.org/10.1176/appi.ajp.2013.12111460.
- van Aken, W. G. (2001). ['Variant of Creutzfeldt-Jacob disease and blood transfusion'; report of the Dutch Health Council]. Ned Tijdschr Geneeskd, 145(30), 1444-1447. ('Variant van de ziekte van Creutzfeldt-Jakob en bloedtransfusie'; rapport van de Gezondheidsraad).
- (Wageningen Bioveterinary research (voorheen Centraal veterinair instituut CVI)). (2023). BSE in Nederland. Retrieved May 2023 from https://www.wur.nl/nl/onderzoek-resultaten/onderzoeksinstituten/biovete….
- Wieser, H. G., Schindler, K., & Zumsteg, D. (2006). EEG in Creutzfeldt-Jakob disease. Clin Neurophysiol, 117(5), 935-951.
- Wilkins, R. H., & Brody, I. A. (1971). Creutzfeldt-Jakob disease. Arch Neurol, 25(6), 572-573. https://doi.org/10.1001/archneur.1971.00490060106011.
- Will, R. G., Ironside, J. W., Zeidler, M., Cousens, S. N., Estibeiro, K., Alperovitch, A., Poser, S., Pocchiari, M., Hofman, A., & Smith, P. G. (1996). A new variant of Creutzfeldt-Jakob disease in the UK. Lancet, 347(9006), 921-925. https://doi.org/10.1016/s0140-6736(96)91412-9.
- Will, R. G., Zeidler, M., Stewart, G. E., Macleod, M. A., Ironside, J. W., Cousens, S. N., Mackenzie, J., Estibeiro, K., Green, A. J., & Knight, R. S. (2000). Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol, 47(5), 575-582.
- Zeidler, M., Stewart, G. E., Barraclough, C. R., Bateman, D. E., Bates, D., Burn, D. J., Colchester, A. C., Durward, W., Fletcher, N. A., Hawkins, S. A., Mackenzie, J. M., & Will, R. G. (1997). New variant Creutzfeldt-Jakob disease: neurological features and diagnostic tests. Lancet, 350(9082), 903-907. https://doi.org/10.1016/s0140-6736(97)07472-2.
- Zerr, I., Kallenberg, K., Summers, D. M., Romero, C., Taratuto, A., Heinemann, U., Breithaupt, M., Varges, D., Meissner, B., Ladogana, A., Schuur, M., Haik, S., Collins, S. J., Jansen, G. H., Stokin, G. B., Pimentel, J., Hewer, E., Collie, D., Smith, P., Sanchez-Juan, P. (2009). Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain, 132(Pt 10), 2659-2668.